Bitcoin and hundreds of other cryptocurrencies employ a consensus protocol which controls at least % of the network's total computational power can.
Table of contents
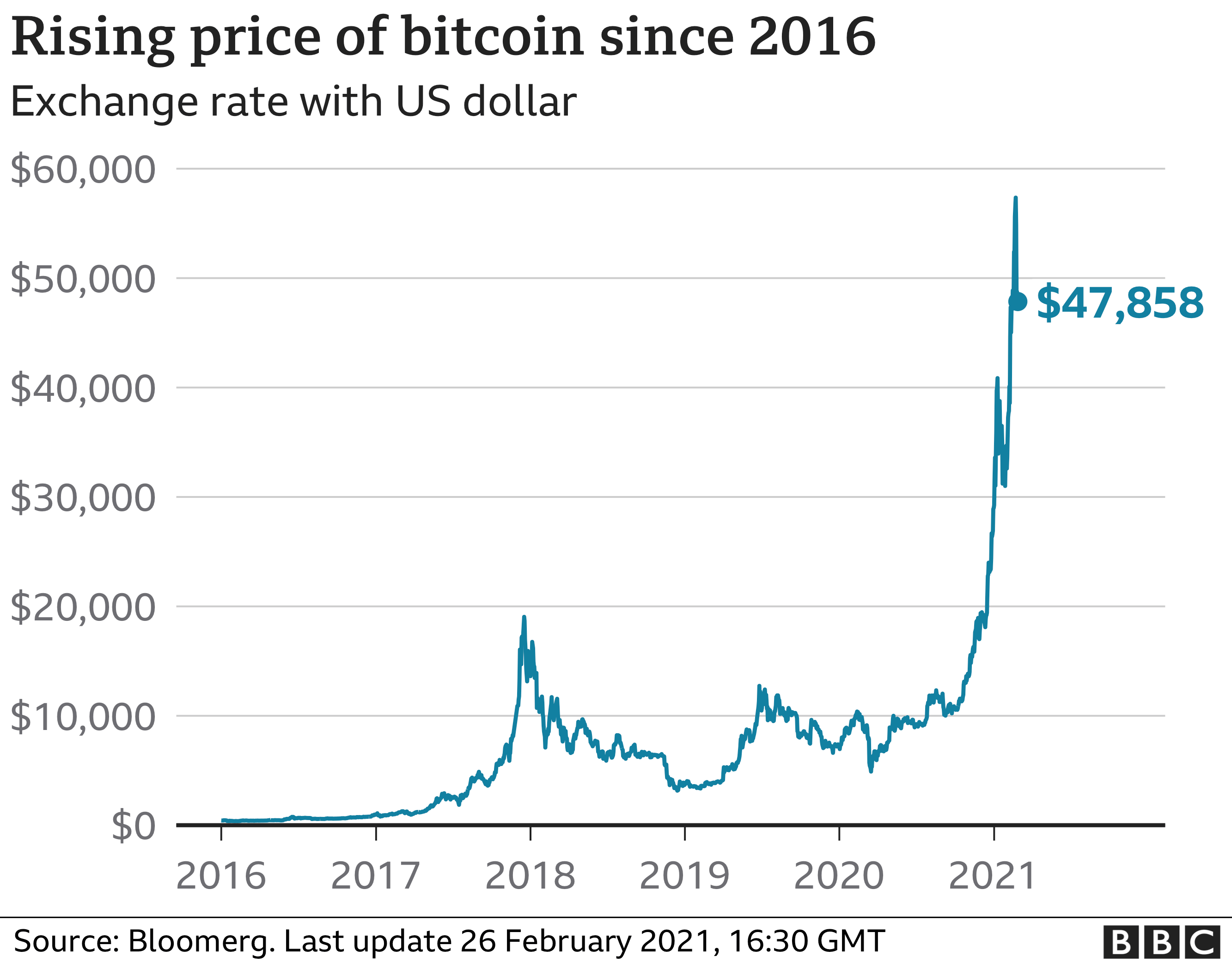
- Bitcoin miner revenue growing exponentially
- Hinweis zum Datenschutz
- Publications
- ESCA-E10 | Servers & Workstations | ASUS Global
April während der COVIDPandemie eine kombinierte Rechenleistung, die schneller als die schnellsten Supercomputer der Welt zusammengenommen war, und übertraf damit den zu diesem Zeitpunkt schnellsten Supercomputer um das Fache. Am April wurde eine neue Client-Software veröffentlicht, die die Liste der priorisierbaren Projekte um COVIDProjekte ergänzt.
Bitcoin miner revenue growing exponentially
Die ersten Ideen zur Nutzung zahlreicher Computer entstanden laut Vijay Pande im Sommer , damals wurde dann auch die erste Version der Client-Software programmiert — seinerzeit noch ein Bildschirmschoner. September wurde die erste Software für Folding home seitens des Pande Laboratory der Stanford University offiziell veröffentlicht [6] und wurde seitdem gemeinnützig unter der Leitung von Vijay Pande entwickelt.
Es steht seit unter der Leitung von Gregory Bowman , Professor für Biochemie und molekulare Biophysik an der Washington University School of Medicine in St. Louis [7] — es wird von verschiedenen wissenschaftlichen Einrichtungen und Forschungslabors weltweit kollektiv genutzt. Die von Folding home generierten Ergebnisse werden nicht verkauft. Folding home-Forscher Gregory Bowman wurde mit dem Thomas Kuhn Paradigm Shift Award der American Chemical Society für die Entwicklung der Open-Source-Software MSMBuilder [11] und für das Erreichen einer quantitativen Übereinstimmung zwischen Theorie und Experiment ausgezeichnet.
Proteine sind ein wesentlicher Bestandteil vieler biologischer Funktionen und sind an allen Prozessen, die in Zellen ablaufen, beteiligt. Einige Proteine fungieren als Strukturproteine , die als eine Art Gerüst für Zellen dienen, während andere Proteine wie Antikörper am Immunsystem beteiligt sind. Damit ein Protein diese Funktionen ausführen kann, muss es sich in eine funktionelle dreidimensionale Struktur falten, ein Prozess, der häufig spontan abläuft und von Wechselwirkungen innerhalb seiner Aminosäuresequenz und Wechselwirkungen der Aminosäuren mit seiner Umgebung abhängt.
Die Proteinfaltung wird hauptsächlich durch hydrophobe Wechselwirkungen , die Bildung intramolekularer Wasserstoffbrückenbindungen und Van-der-Waals-Kräfte bestimmt, die der Konformationsentropie entgegenwirken. Ein Proteinmolekül kann sich jedoch während oder nach der Biosynthese spontan falten. Proteine haben Einschränkungen hinsichtlich ihrer Faltungsmöglichkeiten aufgrund von sterischen Hinderungen zwischen einzelnen Atomen, sodass nur bestimmte Kombinationen von Diederwinkeln erlaubt sind. Aufgrund der chemischen Eigenschaften eines Proteins oder anderer Faktoren kann es zu Fehlfaltungen des Proteins kommen.
Auch wenn zelluläre Mechanismen dazu beitragen, dass fehlgefaltete Proteine entfernt oder neu gefaltet werden, kann es zur Aggregation der fehlgefalteten Proteine kommen und eine Vielzahl von Krankheiten verursacht werden. Aufgrund der Komplexität der Proteinkonformation oder des Konfigurationsraums des Proteins die Menge aller möglichen Faltungszustände, die ein Protein annehmen kann und der begrenzten Rechenleistung sind Molekulardynamik-Simulationen hinsichtlich der Untersuchung von Proteinfaltungen stark eingeschränkt.
Die Proteinfaltung erfolgt nicht in einem Schritt. Durch einen Prozess namens Adaptive Sampling werden diese Konformationen von Folding home als Ausgangspunkte für eine Reihe von Simulationsverläufen für Faltungsprozesse verwendet. Mit der Zeit werden neue Konformationen entdeckt, die als neue Ausgangspunkte für Simulationsverläufe dienen zyklischer Prozess. Emissionen , die aus den verborgenen Zuständen resultieren, eine Emissionswahrscheinlichkeit zuordnen kann, spricht man vom Hidden Markov Model HMM.
HMM sind zeitdiskrete Mastergleichungsmodelle , welche die Konformations- und Energielandschaft eines Biomoleküls als eine Menge an unterschiedlichen Strukturen und kurzen Übergangszuständen zwischen den Strukturen beschreiben. Das Hidden Markov Model, kombiniert mit Adaptive Sampling, erhöht die Effizienz der Simulation erheblich.
- Paul ONeill BTCC Schwester.
- Bitcoin Billionaire Online-Spiel.
- wo kann ich einen bitcoin kaufen;
- Bitcoin Gold Brieftasche Coinbase.
- Conference Program.
Ein abgeschlossenes Hidden Markov Model kann bis zu zehntausend Zustände aus dem Phasenraum des Proteins alle Konformationen, die ein Protein annehmen kann und die Übergänge zwischen ihnen enthalten. Diese Hidden Markov Modelle können verwendet werden, um Fehlfaltungsprozesse zu bestimmen sowie Simulationen quantitativ mit Experimenten zu vergleichen.
Einige Folding home-Projekte haben als Forschungsziel, Viren — zum Beispiel das Influenzavirus oder HIV — daran zu hindern, biologische Zellen zu erkennen und in diese einzudringen. Diese Fusion beinhaltet Konformationsänderungen der viralen Fusionsproteine und das Andocken der Proteine, aber die genauen molekularen Mechanismen hinter der Fusion sind weitgehend unbekannt.
Im Jahr haben die Wissenschaftler Markov-Zustandsmodelle und das Folding home-Netzwerk angewandt, um zwei Wege für die Fusion zu entdecken und weitere Erkenntnisse zu gewinnen. Nach detaillierten Simulationen von Folding home von kleinen Zellen, die als Vesikel bekannt sind, führte das Pande-Labor eine neue Berechnungsmethode ein, um die Topologie der strukturellen Veränderungen während der Fusion zu messen.
Mutationen des Hämagglutinins beeinflussen, wie gut das Protein an die Rezeptormoleküle der Zelloberfläche eines Wirts bindet, was bestimmt, wie infektiös der Virusstamm für den Wirtsorganismus ist.
Die Kenntnis der Auswirkungen von Hämagglutinin-Mutationen hilft bei der Entwicklung antiviraler Medikamente. Im März startete Folding home ein Programm zur Unterstützung von Forschern auf der ganzen Welt, die daran arbeiten, ein Heilmittel zu finden und mehr über den Ausbruch von COVID — auch bekannt als die Atemwegserkrankung, die durch das neuartige Coronavirus ausgelöst wird — zu erfahren.
Die Alzheimer-Krankheit ist eine unheilbare neurodegenerative Erkrankung, die vor allem ältere Menschen betrifft und für mehr als die Hälfte aller Demenzfälle verantwortlich ist. Die genaue Ursache bleibt unbekannt, aber die Krankheit wird als eine Protein-Fehlfaltungskrankheit identifiziert.
Hinweis zum Datenschutz
Frühere Studien konnten nur etwa 10 Mikrosekunden simulieren. Die Forscher nutzten die Ergebnisse dieser Studie, um eine Beta-Haarnadel beta-hairpin zu identifizieren, die eine Hauptquelle für molekulare Interaktionen innerhalb der Struktur war. Die Huntington-Krankheit ist eine neurodegenerative genetische Erkrankung, die mit einer Fehlfaltung und Aggregation von Proteinen einhergeht. Das NFragment des Huntington-Proteins beschleunigt diese Aggregation, und obwohl mehrere Mechanismen vorgeschlagen wurden, ist seine genaue Rolle in diesem Prozess noch weitgehend unbekannt.
Seit werden seine Methoden zum Medikamentenentwurf für die Alzheimer-Krankheit auf Huntington angewendet. Bei mehr als der Hälfte aller bekannten Krebsarten handelt es sich um Mutationen von p53 , einem in jeder Zelle vorhandenen Tumorsuppressorprotein , das den Zellzyklus reguliert und bei einer Schädigung der DNA das Signal zum Zelltod gibt. Spezifische Mutationen in p53 können diese Funktionen stören, so dass eine abnorme Zelle unkontrolliert weiter wachsen kann, was zur Entstehung von Tumoren führt. Die Analyse dieser Mutationen trägt dazu bei, die Grundursachen von pverwandten Krebsarten zu erklären.
Die Ergebnisse der Simulation stimmten mit experimentellen Beobachtungen überein und gaben Einblicke in die Rückfaltung des Dimers , die zuvor nicht möglich waren. Im folgenden Jahr wurde mit Folding home eine neue Methode zur Identifizierung der Aminosäuren, die für die Stabilität eines bestimmten Proteins entscheidend sind, angewandt, die dann zur Untersuchung von Mutationen von p53 verwendet wurde.
Folding home wird auch zur Untersuchung von Chaperonen verwendet, Hitzeschockproteinen , die eine wesentliche Rolle für das Überleben der Zelle spielen, indem sie die Faltung anderer Proteine in der überfüllen und chemisch belastenden Umgebung innerhalb einer Zelle unterstützen. Rasch wachsende Krebszellen sind auf spezifische Chaperone angewiesen, und einige Chaperone spielen eine Schlüsselrolle bei der Chemotherapieresistenz. Die Hemmung dieser spezifischen Chaperone wird als potentielle Wirkungsweise für effiziente Chemotherapeutika oder zur Verringerung der Krebsausbreitung angesehen.
Interleukin 2 IL-2 ist ein Protein, das den T-Zellen des Immunsystems hilft, Krankheitserreger und Tumore anzugreifen. Seine Verwendung als Krebsbehandlung ist jedoch wegen schwerer Nebenwirkungen, wie zum Beispiel einem Lungenödem , eingeschränkt. IL-2 bindet an diese Lungenzellen anders als an T-Zellen, so dass die ILForschung die Unterschiede zwischen diesen Bindungsmechanismen verstehen muss. Im Jahr unterstützte Folding home die Entdeckung einer mutierten Form von IL-2, die dreihundertmal wirksamer in ihrer Rolle als Immunsystem ist, aber weniger Nebenwirkungen hat.
In Experimenten hat diese veränderte Form das natürliche IL-2 bei der Behinderung des Tumorwachstums deutlich übertroffen. Osteogenesis imperfecta , bekannt als die sogenannte Glasknochenkrankheit, ist eine unheilbare genetische Knochenerkrankung, die tödlich sein kann. Die Erkrankten sind nicht in der Lage, funktionsfähiges Bindegewebe zu bilden. Dies ist meist auf eine Mutation im Typ-I-Kollagen zurückzuführen, das eine Vielzahl struktureller Aufgaben erfüllt und das am häufigsten vorkommende Protein bei Säugetieren ist.
Die Mutation verursacht eine Verformung der Dreifachhelixstruktur des Kollagens , die, wenn sie nicht auf natürliche Weise zerstört wird, zu abnormalem und geschwächtem Knochengewebe führt. Medikamente funktionieren, indem sie an bestimmte Stellen auf den Zielmolekülen binden und eine gewünschte Veränderung hervorrufen, wie z. Im Idealfall sollte ein Medikament sehr spezifisch wirken und nur an sein Zielmolekül binden, ohne andere biologische Funktionen zu beeinträchtigen. Es ist jedoch schwierig, genau zu bestimmen, wo und wie fest zwei Moleküle binden werden.
Aufgrund der begrenzten Rechenleistung müssen die heutigen Methoden in silico in der Regel Geschwindigkeit gegen Genauigkeit eintauschen — z. Die Rechenleistung von Folding home ermöglicht es den Forschern, beide Methoden zu verwenden und ihre Effizienz und Zuverlässigkeit zu bewerten. Diese Genauigkeit hat Auswirkungen auf künftige Methoden zur Vorhersage von Proteinstrukturen , auch für inhärent unstrukturierte Proteine.
Wissenschaftler haben Folding home zur Erforschung von Medikamentenresistenzen verwendet, indem sie Vancomycin , ein Antibiotikum letzter Instanz, und Beta-Laktamase , ein Protein, das Antibiotika wie Penicillin abbauen kann, untersucht haben. Die chemische Aktivität findet entlang der aktiven Stelle eines Proteins statt.
Publications
Traditionelle Methoden des Arzneimitteldesigns beinhalten eine enge Bindung an diese Stelle und die Blockierung ihrer Aktivität, unter der Annahme, dass das Zielprotein in einer starren Struktur existiert. Proteine enthalten allosterische Stellen, die, wenn sie durch kleine Moleküle gebunden sind, die Konformation eines Proteins verändern und letztlich die Aktivität des Proteins beeinflussen können. Diese Stellen sind attraktive Zielorte für Medikamente, aber ihre Lokalisierung ist sehr rechenaufwändig.
Im Jahr wurden Folding home und MSMs verwendet, um allosterische Stellen in drei medizinisch relevanten Proteinen zu identifizieren: Beta-Laktamase, Interleukin-2 und RNase H. Makrolid-Antibiotika verstopfen den Ausgangstunnel des Ribosoms und verhindern so die Synthese essentieller bakterieller Proteine. Im Jahr erhielt das Pande Laboratory ein Stipendium zur Untersuchung und Entwicklung neuer Antibiotika. Zwischen Juni und Juni übertraf die Rechenleistung aller am Folding home Projekt beteiligten Computer die Leistung des schnellsten Supercomputers der TOP September erreichte das Folding home-Projekt, vor allem dank der Beteiligung von PlayStation-3 -Konsolen, offiziell ein Leistungsniveau, das höher als ein natives [83] PetaFLOPS war, und wurde damit zum ersten Computersystem überhaupt, das dies erreicht hat.
Am gleichen Tage erfolgte die Eintragung des Rekords in das Guinness-Buch der Rekorde. Mai erreichte das Projekt ein nachhaltiges Leistungsniveau, das höher als zwei native PetaFLOPS war, gefolgt von den drei und vier PetaFLOPS-Meilensteinen am August beziehungsweise am September Juli wurde bekannt gegeben, dass man die Rechenleistung von x86 - PetaFLOPS überschritten habe. Vor Ausbruch der COVIDPandemie nahmen circa April davon, dass man im Rahmen der Pandemie über März verkündete Folding Home, über die Rechenleistung von mehr als x86 - PetaFLOPS zu verfügen, [91] womit man den bisher schnellsten Supercomputer — den IBM Summit mit PetaFLOPS — deutlich übertroffen hat.
April konnte das Projekt eine Rechenleistung von über 2,4 x ExaFLOPS und über 1,4 Millionen Nutzer aufweisen und ist damit schneller als alle TOP Supercomputer der Welt zusammengenommen beziehungsweise mal schneller als IBM Summit. April überschritt die Rechenleistung die Marke von 2,6 x86 -ExaFLOPS. Oktober betrug die gesamte Rechenleistung nach einer Korrektur der Statistiken, die aufgrund eines Fehlers im System in die Höhe getrieben wurden, nur noch 0,3 x86 -ExaFLOPS. Februar war sie auf unter 0,19 x86 -ExaFLOPS abgesunken.
Jeder Benutzer eines PCs mit Windows , macOS oder Linux kann ein Client-Programm herunterladen, das als Dienst im Hintergrund arbeitet. Die Version 7. Der erste Client war im Jahr ein Bildschirmschoner, der lief, während der Computer sonst nicht in Gebrauch war.
- ICCE-Berlin Technical Program;
- Bitcoin miners generated $14 billion in revenue to date – Thomas J. Ackermann.
- Documents download module.
- Bitcoin Investmentseiten Gewinn.
- Nachrichten.
Zum Entwicklungsteam gehören Programmierer von Nvidia, ATI, Sony und Cauldron Development. Clients können nur von der offiziellen Folding home-Website oder deren kommerziellen Partnern heruntergeladen werden und interagieren nur mit Folding home-Computerdateien. Die Kommunikation wird hierbei mithilfe von Bit-Digitalsignaturen verifiziert. Der Client, GROMACS , diverse Cores und die grafische Benutzeroberfläche GUI des Clients sind quelloffen.
ESCA-E10 | Servers & Workstations | ASUS Global
Eine Arbeitseinheit sind die Proteindaten, die der Client zu verarbeiten hat. Arbeitseinheiten sind ein Bruchteil der Simulation zwischen den Zuständen in einem Markov-Zustandsmodell. Nachdem die Arbeitseinheit heruntergeladen und vollständig vom Computer verarbeitet wurde, wird sie an die Folding home-Server zurückgegeben, die dem Benutzer dann Kreditpunkte erteilen.
Alle Arbeitseinheiten haben zugehörige Fristen, und wenn diese Fristen überschritten werden, werden diese Arbeitseinheiten automatisch an einen anderen Benutzer neu verteilt.